|
|
|
|||

| Распечатать |
Синтез и биологическая активность бензимидазофталазинов

УДК 576.89
DOI 10.33861/2071-8020-2026-2-26-29
Оригинальное эмпирическое исследование
Фетисов Л. Н., Зубенко А. А., Святогорова А. Е., Яровая Н. А. Северо-Кавказский зональный научноисследовательский
ветеринарный институт – филиал Федерального государственного бюджетного научного учреждения
«Федеральный Ростовский аграрный научный центр», г. Новочеркасск
Диваева Л. Н. Научно-исследовательский институт физической и органической химии Южного Федерального университета,
г. Ростов-на-Дону
Аннотация. Фталазин определяется как органическое гетероциклическое соединение, также известное как бензопиридазин или бензоортодиазин, которое обладает структурной универсальностью, что повышает его эффективность в различных биологических областях, включая противоопухолевые и антимикробные свойства. Целью исследований был синтез конденсированных производных фталазина, а именно, бензимидазофталазинов. Взаимодействием замещённого фталазона 1 с 2,4-динитрохлорбензолом 2 в диме-тилформамиде в присутствии карбоната калия получен промежуточный продукт 3, который последовательным восстановлением железом в соляной кислоте и термической циклизацией в присутствии минеральных кислот был превращён в аминопроизводное 4. Ацилированием соединения 4 ацилгалогенидами был получен ряд конденсированных производных фталазина. In vitro изучена биологическая активность 16 синтезированных соединений 1А-16А в отношении простейших вида Colpoda steinii, в отношении микроскопических грибов вида Penicillium italicum, в отношении грамположительных бактерий вида Staphylococcus aureus VKM B-128, в отношении грамотрицательных бактерий вида Esherichia coli VKM B-825. Установлено, что 8 синтезированных соединений бензими-дазофталазина (1А, 2А, 3А, 9А, 10А, 14А, 15А, 16А) обладают антибактериальной активностью в отношении Staphylococcus aureus на уровне 42,8-57,1% активности препарата сравнения ципрофлоксацина (антибиотик ряда фторхинолонов) и 60 - 80 % активности препарата сравнения фуразолидона (антибактериальный нитрофурановый препарат). В отношении Esherichia coli антибактериальная активность изученных соединений была ниже и составляла 18,4-26,3% активности ципрофлоксацина и 38,8-55,5% активности фуразо-лидона. То есть, грамположительные бактерии более чувствительны к синтезированным соединениям. Таким образом, открыта новая группа соединений с перспективой использования в качестве антибактериальных средств.
Ключевые слова: фталазин, бензимидазол, бензимидазофталазины, биологически активные конформации, гибриды фталгидразида и бензимидазола, антибактериальная активность, ципрофлоксацин, фуразолидон
Фталазин, также известный как бензопиридазин или бензоортодиазин, обладает структурной универсальностью, что повышает эффективность различных фармацевтических препаратов на его основе, включая противоопухолевую и антимикробнную активности.
Фталазин-1(2Н)-он (фталазинон) представляет собой гетероцикл, имеющий таутомеризуемое 1,2-диазиновое кольцо, которое не является обычным структурным элементом в природных продуктах. Однако этот бензодиазин представляет собой интересную основу для создания лекарственных средств и полимерных материалов. Следует отметить универсальность этой гетеробициклической системы в области разработки лекарственных средств, поскольку она способна взаимодействовать с различными видами биологических мишеней, имеющих отношение к широкому спектру патологических процессов, включая диабет, рак, астму, аллергии, боль и воспаление, а также инфекционные, сердечно-сосудистые или неврологические заболевания. В последние годы производные фталазина стали предметом обширных исследований. Среди этих фталазинов 4-замещенные фталазины действуют как эффективные противовоспалительные, противомикробные и противоопухолевые соединения. Кроме того, производные 1-хлорфталазина считаются важными компонентами в конструкции биологически активных молекул, таких как тетразол, триазол, имидазол, пиримидин, пиперазин и триазин, которые обладают замечательной противоопухолевой, противомалярийной и антагонистической активностью в отношении андрогенных рецепторов [1, 4]. Кроме того, ряд производных 2,3-ди-гидрофталазин-1,4-диона хорошо известны как активные противосудорожные средства.
Конденсированные производные фталазина также широко представлены в литературе.
В данной работе Karina Gonzalez et all (2024) установили у 3-(арил)-6-пиперазин-1,2,4-триазоло[3,4-а]фталазинов их большую эффективность в качестве лейшманицидных средств при использовании в виде наночастиц или микрочастиц со средними размерами 250, 400, 600-900 или 900-2000 нм, инкапсулированными в полимерную матрицу [14].
Hatem Hussein Bayoumi et all (2024) разработали, синтезировали и оценили новые производные фталазина в отношении линий раковых клеток печени G2 и MCF-7 и в качестве ингибиторов VEGFR-2 (фактор роста эндотелия сосудов). В частности, было обнаружено, что соединения 2g и 4a являются наиболее эффективными производными среди всех протестированных соединений против линий раковых клеток MCF-7 и Hep G2 со значениями IC50 0,15 и 0,12, а также 0,18 и 0,09 мкм (микромоль) соответственно. В качестве препарата сравнения в данном исследовании использовался сорафениб. Были проведены исследования по молекулярному моделированию всех соединений с активным центром VEGFR-2. Данные, полученные в результате биологических испытаний, в значительной степени коррелировали с данными, полученными в результате молекулярного моделирования. Более того, результаты молекулярного докинга (MD) показали стабильность взаимодействия лиганд–мишень. Кроме того, производные 2g и 4a показали хороший профиль ADMET, рассчитанный in silico [12].
Krishnappa B. Badiger et all (2024) описали быстрый и недорогой однокомпонентный и многокомпонентный синтез 1Н-пира-золо[1,2-b]фталазин-5,10-диона путем реакции конденсации ари-лальдегида, малононитрила или этилцианацетата и фталгидразида, катализируемой водным экстрактом золы коры папайи (WEPBA) в среде растворителя при микроволновом облучении. Используемая каталитическая среда безвредна для окружающей среды и обладает рядом преимуществ: она проста, недорога, даёт высокие выходы, проста в обработке и не требует использования опасного растворителя для проведения реакции. Некоторые из выбранных производных 1Н-пиразоло[1,2-b]фталазин-5,10-диона были оценены in vitro на антиоксидантную активность с использованием метода DPPH-анализа. Ряд соединений обладают значительными антиоксидантными свойствами по сравнению с эталонной аскорбиновой кислотой [15].
В работе Huan He et all (2023) описаны полициклические (тетрациклические) производные фталазина. Отмечено, что мутации белка опухолей KRAS (G12C, G12D) участвуют в онкогенезе и прогрессировании многих наиболее смертоносных видов рака. Один из семи гомологов (SOS1) является важнейшим регулятором KRAS, который переводит KRAS из неактивного состояния в активное. В этой работе авторы сообщают о синтезе производных тетрациклического фталазина для избирательного ингибирования SOS1 в отношении EGFR. Ведущее соединение 6c продемонстрировало замечательную активность в ингибировании пролиферации клеток поджелудочной железы, мутантных по KRAS(G12C), а также 6с продемонстрировал благоприятный фармакокинетический профиль in vivo с биодоступностью 65,8% и продемонстрировал мощное подавление опухоли на моделях ксенотрансплантатов опухоли поджелудочной железы. Эти интригующие результаты позволяют предположить, что 6c потенциально может быть использован в качестве лекарственного средства для лечения опухолей, вызываемых KRAS [13].
Бычий сывороточный альбумин был использован Alireza Farahanipour et all (2024), в качестве экологически чистого, недорогого и эффективного биокатализатора для синтеза производных
1Н-пиразоло[1,2-b]фталазин-5,10-диона и 1Н-пиразоло[1,2-a]пири-дазин-5,8-диона посредством трехкомпонентной реакции с хорошим или отличным выходом (73-93%). К замечательным особенностям этого подхода относятся экологичный катализатор, отсутствие органических растворителей, отсутствие металлов, мягкая реакция, простота обработки и устойчивость функциональных групп в условиях реакции [5].
Фармакологическое ингибирование онкогенных протеинкиназ с помощью небольших молекул является многообещающей стратегией борьбы с несколькими злокачественными новообразованиями человека. CDK1 (фермент, циклин-зависимая киназа) является примером такой ценной мишени для лечения протоковых аденокарцином поджелудочной железы (PDAC); его сверхэкспрессия в PDAC положительно коррелирует с размером, гистологической оценкой и агрессивностью опухоли. Laila Akl et all (2022) сообщают о синтезе новой серии производных 1-пиперазинил-4-бензилфталазина (8a-g, 10a-i и 12a-d) в качестве перспективных противоопухолевых средств с ингибирующей активностью CDK1. Антипролиферативная активность этих препаратов была сначала исследована на панели из 11 клеточных линий, представляющих 5 видов рака (поджелудочной железы, меланомы, лейкемии, толстой кишки и молочной железы), а затем подтверждена на двухклеточных линиях PDAC, сверхэкс-прессирующих CDK1 (клетки MDA-PATC53 и PL45). Фталазины 8g, 10d и 10h проявляли сильную активность в отношении клеточных линий MDA-PATC53 (IC50 = 0,51, 0,88 и 0,73 мкм соответственно) и PL45 (IC50 = 0,74, 1,14 и 1,00 мкм соответственно). Кроме того, соединения 8g, 10d и 10h проявляли мощную и избирательную ингибирующую активность в отношении CDK1 с IC50 в диапазоне 36,80–44,52 нМ. В заключение, была выявлена группа мощных и селективных ингибиторов CDK1, которые могут служить ведущими соединениями для разработки дальнейших ингибиторов CDK1 [16].
С применением карбоксиметилцеллюлозы в качестве реакционной среды Farzaneh Mohamadpour (2022) описан экологически безопасный способ получения производных 1Н-пиразоло [1,2-b] фталазин-5,10-диона путем циклоконденсации Кневенагеля-Михаэ-ля, основанный на принципах «зеленой химии». Основные преимущества включают использование пригодного для вторичной переработки экологически чистого катализатора, доступные в продаже недорогие исходные материалы, простоту эксплуатации, безопасные условия проведения реакции, высокую атомарную экономичность, отсутствие растворителей, короткое время реакции и высокие выходы [11].
Разработка гибридных молекул с двойными функциями является одним из подходов к повышению терапевтической эффективности комбинированного лечения. Ранее Tai-Lin Chen et all (2021) конъюгировали фармакофоры фталазина и бис(гидроксиметил)пир-рола для получения гибридов, обладающих антиангиогенетической активностью и активностью к сшиванию ДНК между нитями. Чтобы улучшить биодоступность, авторы применили бензологический подход к разработке и синтезу новой серии 1,2-бис(гидроксиметил) бензо[g]пирроло[2,1-a]фталазинов. Эти новые гибриды сохранили двойную функцию и могут быть изготовлены в виде носителей для внутривенного и перорального введения. Среди них соединение 19а с диметиламиногруппой в положении С6 заметно подавляло рост опухолей линии клеток мелкоклеточного рака легких человека H526, линии клеток плоскоклеточного рака легких H520 и линии клеток рака почки 786-O у мышей nude, подразумевая, что соединение 19а является противоопухолевым средством широкого спектра действия. Авторы показали, что сочетание антиангиогенного действия и перекрестного связывания ДНК, вероятно, является полезным подходом к повышению эффективности комбинированной терапии [22].
Anupreet Kharbanda et all (2021) исследовали ряд фталазинов на предмет воздействия на путь TGF? с помощью нерецепторно-ки-назного механизма. Соединение (10р) продемонстрировало многообещающее ингибирование пути TGF? при IC50 0,11 ± 0,02 мкм с высоким показателем селективности. Селективный скрининг выявил ингибирование пути TGF? по нерецепторно-киназному механизму [6].
Противоопухолевая активность ряда дифункциональных замещенных 1,2-дигидрофталазинов была изучена Lyudmyla M. Potikha et all (2022) в рамках международной научной программы «Скрининг линий опухолевых клеток человека NCI-60». Скрининг был проведен in vitro на 60 клеточных линиях легких, почек, ЦНС, яичников, предстательной железы и молочной железы, эпителиального рака, лейкемии и меланомы. Наиболее эффективными оказались соединения с арильным заместителем в N-2 звене фталазинового цикла: 4-(4-хлорфенил)-2-(4-метилфенил)-1,2-дигидрофталазин (средний показатель lg GI50 = -5,79, lg TGI = -5,42, lg LC50 = -4,97) и 2,4-бис(4-хлорфенил)-1,2-дигидрофталазин (средний показатель LGI50 = -5,57, lg TGI = -5,08, lg LC50 = -4,61) [17].
Новые связанные с фталазоном производные 1,2,3-триазола 12-21 были синтезированы Mohamed A. Abdelgawad et all (2023) с использованием катализируемых Cu(I) реакций алкинфунк-ционализированного фталазона 1 с функционализированными азидами. Антипролиферативная оценка производных показала высокую активность ряда соединений по сравнению с противоопухолевым препаратом доксорубицином. Производные 16, 18 и 21 были оценены на предмет ингибирующей активности VEGFR-2, и в результате оказалось, что производное 16 проявило более высокую активность по сравнению с сорафенибом. Соединение 16 вызывало нарушение распределения MCF7 по клеточному циклу и увеличивало процент клеток в S-фазе в 1,37 раза. Молекулярный докинг in silico эффективных производных 16, 18 и 21 против рецептора фактора роста эндотелия сосудов-2 (VEGFR-2) подтвердил формирование стабильных белково–лигандных взаимодействий внутри кармана [18].
В результате взаимодействия фталгидразида с соответствующими производными бромпропоксикумарина Dusica Simijonovic et all (2021) был получен ряд новых гибридов фталгидразида и кумарина. Эти реакции проводили в присутствии Cs2CO3, в ацетонитриле в качестве растворителя и при кипячении в течение 5 ч. Полученные гибриды содержат один или два кумариновых каркаса, связанных с фталгидразидным азотом с помощью про-поксиллинкера. Были получены десять новых гибридов фталгидразида и кумарина с умеренным или хорошим выходом. Прогноз потенциальной биологической активности синтезированных соединений был выполнен с помощью онлайн-программы PASS (прогнозирование спектров активности веществ). На основании полученных результатов для исследования молекулярного докинга был выбран рецептор серотонина 2A (5-HT2AR). Также был проведен молекулярный докинг с коммерчески доступным препаратом Рисперидоном. Полученные результаты были сопоставлены с потенциальными биологически активными конформациями полученных гибридов фталгидразида и кумарина [10].
Целью наших исследований был синтез конденсированных производных фталазина, а именно, бензимидазофталазинов общей формулы А (рисунок 1).
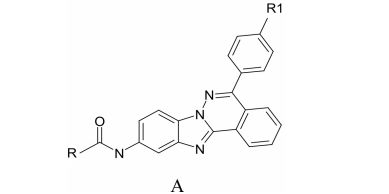
Рис. 1. Бензимидазофталазины общей формулы А
Материалы и методы исследований. Синтез соединений был произведен согласно схеме, представленной на рисунке 2.
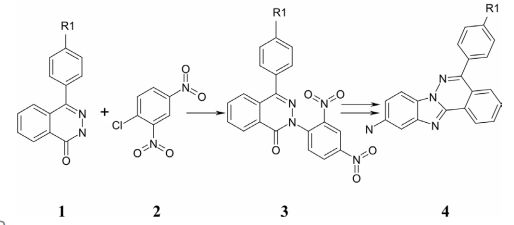
Рис. 2. Синтез соединений
Взаимодействием замещённого фталазона 1 с 2,4-динитрох-лорбензолом 2 в диметилформамиде в присутствии карбоната калия нами получен промежуточный продукт 3, который последовательным восстановлением железом в соляной кислоте и термической циклизацией в присутствии минеральных кислот был превращён в аминопроизводное 4. Ацилированием соединения 4 ацилгалогенидами был получен ряд производных общей формулы А (рисунок 1). Номера соединений и значения радикалов R и R1 представлены в таблице 1.
Таблица 1 Номера соединений и значения радикалов R и R1
|
Номера соединений |
R |
R1 |
|---|---|---|
|
1А |
метил |
хлор |
|
2А |
2,4-диметилпиразолилметил |
хлор |
|
3А |
орто-метоксифенил |
хлор |
|
4А |
пара-нитрофенил |
хлор |
|
5А |
трет-бутил |
хлор |
|
6А |
орто-хлорфенил |
хлор |
|
7А |
этил |
хлор |
|
8А |
фенил |
хлор |
|
9А |
фенил |
метил |
|
10А |
пара-толилсульфонил |
метил |
|
11А |
ацетаминофенилметил |
метил |
|
12А |
2-фенилвинил |
метил |
|
13А |
2-метоксифенил |
метил |
|
14а |
2,4-диметилпиразолилметил |
метил |
|
15А |
орто-хлорфенил |
метил |
|
16А |
2,4-динитрофенил |
метил |
Антибактериальную активность в отношении E.coli и St.aureus, фунгистатическую активность в отношении P.italicum определяли диско-диффузионным способом [2, 8, 9, 20, 21]. Протистоцидная активность изучалась методом серийных разведений [3, 8, 19].
Результаты исследований и их обсуждение. Биологическая активность соединений 1А-16А представлена в таблице 2.
Таблица 2 Биологическая активность новых соединений 1А-16А
|
№ соединений |
Colpoda steinii., мкг/мл |
Penicillium italicum, мм |
Staphylococc. aureus VKM B-128, мм |
Esherichia coli VKM B-825, мм |
|---|---|---|---|---|
|
1А |
>500 |
0 |
15 |
7 |
|
2А |
>500 |
0 |
17 |
7 |
|
3А |
>500 |
0 |
20 |
7 |
|
4А |
>500 |
0 |
8 |
7 |
|
5А |
>500 |
0 |
10 |
7 |
|
6А |
>500 |
0 |
10 |
7 |
|
7А |
>500 |
0 |
13 |
7 |
|
8А |
>500 |
0 |
13 |
7 |
|
9А |
>500 |
0 |
17 |
7 |
|
10А |
>500 |
0 |
15 |
7 |
|
11А |
>500 |
0 |
7 |
7 |
|
12А |
>500 |
0 |
9 |
7 |
|
13А |
>500 |
0 |
12 |
7 |
|
14А |
>500 |
0 |
15 |
10 |
|
15А |
>500 |
0 |
17 |
10 |
|
16А |
>500 |
0 |
17 |
10 |
|
Ципрофлоксацин |
|
|
35 |
38 |
|
Фуразолидон |
|
|
25 |
18 |
|
Фундазол |
|
50 |
|
|
|
Хлорохин |
3,9 |
|
|
|
Из данных таблицы 2 следует, что 8 синтезированных соединений бензимидазофталазина (1А, 2А, 3А, 9А, 10А, 14А, 15А, 16А) обладают антибактериальной активностью в отношении Staphylococcus aureus на уровне 42,8-57,1% активности препарата сравнения ципрофлоксацина (антибиотик ряда фторхинолонов) и 60-80% активности препарата сравнения фуразолидона (антибактериальный нитрофурановый препарат). В отношении Esherichia coli антибактериальная активность изученных соединений была ниже и составляла 18,4-26,3% активности ципрофлоксацина и 38,8-55,5% активности фуразолидона. То есть, грамположительные бактерии более чувствительны к синтезированным соединениям. Таким образом, открыта новая группа соединений с перспективой использования их в качестве антибактериальных средств. В итоге, получены биологически активные конформации гибридов фталгидразида и бензимидазола.
Список литературы:
1. Зубенко А. А., Фетисов Л. Н., Святогорова А. Е. [и др.]. Биологическая активность сульфаниламидов фталазина. Ветеринария и кормление. 2025; (6): 38-42.
2. Бовкунова А. А., Бажина Е. С., Шмелев М. А. [и др.]. Новые координационные соединения марганца(II) с 4-((1H-пиррол-2-ил)метилен-ами-но)-4H-1,2,4-триазолом. Координационная химия. 2025; (51 (7): 464-484.
3. Фетисов Л. Н., Зубенко А. А., Святогорова А. Е. [и др.]. Простые объективные и информативные методики скрининга новых соединений с биологической активностью с целью отбора активно действующих субстанций для ветеринарной медицины. Ветеринария Северного Кавказа. 2025; (11): 409-425.
4. Зубенко А. А., Фетисов Л. Н., Святогорова А. Е. [и др.]. Синтез и биологическая активность триазолофталазинов. Ветеринария Кубани. 2025; (1): 28-31.
5. Alireza Farahanipour, Hossein Bavandi, Mansour Shahedi, Zohreh Habibi. Polycyclic Aromatic Compounds: Synthesis of 1H-Pyrazolo[1,2-b] Phthalazine-5,10-Dione and 1H-Pyrazolo[1,2-a]Pyridazine-5,8-Dione Derivatives by Bovine Serum Albumin in Water. 2023; (43 (8): 7042-7051.
6. Anupreet Kharbanda, Lingtian Zhang, Debasmita Saha, Phuc Tran, Ke Xu, Ming O. Li, Yuet-Kin Leung, Brendan Frett, Hong-yu Li. European Journal of Medicinal Chemistry: Discovery and biological evaluation of phthalazines as novel non-kinase TGF? pathway inhibitors. 2021; (223): 113660.
7. Ermakova E. A., Golubeva Yu. A., Smirnova K. S. et al. Bioactive mixed-ligand zinc(II) complexes with 1H-tetrazole-5-acetic acid and oligopyridine derivatives. Polyhedron. 2023; (230): 116213.
8. Matiukhina A. K., Vladimirova А. E., Zorina-Tikhonova Е. N. et al. Complexes of Mn(II) and Сo(III) with 2-Amino-N'-(pyridin-2-ylmethylene) benzohydrazide: Synthesis, Structure, and In Vitro Biological Activity. Russian Journal of General Chemistry. 2023; (93 (S2): S596-S604.
9. Ermakova E. A., Golubeva Yu. A., Smirnova K. S. et al. Cytotoxic mixed-ligand copper(ii) complexes with 1H-tetrazole-5-acetic acid and oligopyridine derivatives. New Journal of Chemistry. 2023; (47 (19): 9472-9482.
10. Simijonovic D., Vlachou E.-E. N., Litinas K. E. et al. Synthesis, structural characterization, and molecular docking study of new phthalhydrazidecoumarin hybrids. Journal of Molecular Structure. 2021; (1226 B): 129366.
11. Mohamadpour F. Carboxymethyl Cellulose (CMC) as a Recyclable Green Catalyst Promoted Eco-Friendly Protocol for the Solvent-Free Synthesis of 1H-Pyrazolo[1,2-b]Phthalazine-5,10-Dione Derivatives. Polycyclic Aromatic Compounds. 2022. (42 (4): 1091-1102.
12. Bayoumi H. H. et al. Exploration of the VEGFR-2 inhibition activity of phthalazine derivatives: design, synthesis, cytotoxicity, ADMET, molecular docking and dynamic simulation. RSC Advances. 2024; (14 (30): 21668-21681.
13. He H. et al. Design of Orally-bioavailable Tetra-cyclic phthalazine SOS1 inhibitors with high selectivity against EGFR. Bioorganic Chemistry. 2023; (136): 106536.
14. Gonzalez K. et al. In vitro anti-trypanosomal activity of 3-(aryl)-6-piperazin1,2,4-triazolo[3,4-a]phthalazines-loaded ultrathin polymeric particles: effect of polymer type and particle size. RSC Pharmaceutics. 2024; (1 (1): 108-120.
15. Krishnappa B. Badiger et al. An Agro-Waste Catalyzed Facile Synthesis of 1H-Pyrazolo[1,2-b]Phthalazine-5,10-Dione Derivatives: Evaluation of Antioxidant and Electrochemical Studies. Polycyclic Aromatic Compounds. 2024; (44 (2): 1128-1150.
16. Laila Akl et al. Identification of novel piperazine-tethered phthalazines as selective CDK1 inhibitors endowed with in vitro anticancer activity toward the pancreatic cancer. European Journal of Medicinal Chemistry. 2022; (243): 114704.
17. Potikha L. M. et al. Anticancer evaluation of difunctional substituted 1,2-dihydrophthalazines. Chemical Data Collections. 2022; (37): 100817.
18. Abdelgawad M. A. et al. Phthalazone tethered 1,2,3-triazole conjugates: In silico molecular docking studies, synthesis, in vitro antiproliferative, and kinase inhibitory activities. Bioorganic Chemistry. 2023; (133): 106404.
19. Divaeva L. N., Zubenko A. A., Morkovnik A. S. et al. Synthesis of New N-[?-(Hetero)arylethyl]benzimidazole-2-carbothioamides and Their Analogues as Anti-Infective Agents and Compounds with Possible Neuro(psycho)tropic and Anticancer activity. Russian Journal of General Chemistry. 2024; (94 (2): 341-351.
20. Baranov V. V., Vikrishchuk N. I., Zubenko A. A. et al. Synthesis, Structure, and Biological Activity of New Metal Carboxylate Complexes Containing a Glycoluril Fragment. Russian Journal of General Chemistry. 2023; (93 (4): 863-869.
21. Ermakova E. A., Golubeva Yu. A., Smirnova K. S. et al. Synthesis, structure and biological properties of the zinc(II) complexes with 5-(4-chlorophenyl)-1H-tetrazole and oligopyridine derivatives. Inorganica Chimica Acta. 2024; (571): 122217.
22. Chen T.-L. et al. Discovery of Oral Anticancer 1,2-Bis(hydroxymethyl) benzo[g]pyrrolo[2,1-a]phthalazine Hybrids That Inhibit Angiogenesis and Induce DNA Cross-Links. Journal of Medicinal Chemistry. 2021; (64 (17): 12469-12486.
Сведения об авторах:
Фетисов Леонид Николаевич, кандидат ветеринарных наук, ведущий научный сотрудник творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-908-1978224; e-mail: fetisoff.leonid2018@yandex.ru.
Зубенко Александр Александрович, доктор биологических наук, главный научный сотрудник творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-928-6049743; e-mail: alexsandrzubenko@yandex.ru.
Яровая Наталья Александровна, лаборант-исследователь творческого коллектива по химическому синтезу новых лекарственных соединений Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-999-2340481; e-mail: tkhim.sintez@yandex.ru.
Диваева Людмила Николаевна, кандидат химических наук, ведущий научный сотрудник научно-исследовательского института физической и органической химии Южного федерального университета; 344090, Ростовская область, г. Ростов-на-Дону, просп. Стачки, 194/2; тел.: 8-928-1756654; e-mail: divaevaln@mail.ru.
Ответственный за переписку с редакцией: Святогорова Александра Евгеньевна, кандидат сельскохозяйственных наук, ведущий научный сотрудник творческого коллектива по химическому синтезу новых лекарственных соединений, ученый секретарь Северо-Кавказского зонального научно-исследовательского ветеринарного института – филиала ФГБНУ «Федеральный Ростовский аграрный научный центр»; 346421, Ростовская область, г. Новочеркасск, Ростовское шоссе, 0; тел.: 8-988-9525755; e-mail: sviatogorova.a@yandex.ru.
Заявленный вклад авторов: рукопись была написана благодаря вкладу всех авторов. Все авторы одобрили окончательную версию рукописи.
Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
| 2026 © Ветеринария Кубани | Разработка сайта - Интернет-Имидж | |
|---|---|---|